
For many years, the perceived wisdom in the medical device sector was to always launch in your home territory first. For UK and EU based companies, this meant obtaining a CE mark and then turning their attention to the US. However, in recent years the delays in medical device regulatory approvals within the EU have prompted UK- and EU-based startups to re-evaluate this approach, with most now opting to pursue approval from the US Food and Drug Administration (FDA) first.
At eg technology, we are seeing an increasing demand for our design services from European companies targeting US regulatory approval. This blog explores some of the reasons behind this trend and discusses why a regulatory strategy prioritising FDA approval is proving advantageous for medical device startups and is increasingly being demanded by investors.
Long lead-times and the relative uncertainty of EU regulatory approvals
It is well publicised that the EU regulatory process for medical devices has undergone significant changes in recent years, with the introduction of the Medical Device Regulation (MDR) and In-Vitro Diagnostic Regulation (IVDR). While these updates to the regulations have brought important improvements, they have also introduced a more rigorous evaluation process. In conjunction with resource/capacity constraints at the notified bodies there has been backlog of applications, creating a significant bottleneck for medical device companies getting their products to market.
This has hit start-ups and SMEs particularly hard, as not only do the associated costs and fees continue to climb, but more critically for SMEs the long-lead time on gaining approvals creates immense funding challenges. There is now routinely a lengthy period, often in excess of 12 months, between completing the design, development and clinical evaluation and actually getting the CE mark, allowing the start of commercial activities.
In addition, the new regulations have brought with them uncertainty regarding interpretation and enforcement of the updated requirements. Examination has certainly become more robust, but the early years of the MDR saw a marked absence of authoritative guidance on the application and interpretation of these new rules. This led to a sense of concern over whether the updated rules were being consistently applied, particularly on lower classification devices where a risk-averse review process was demanding significantly increased levels of evidence.
As a result, many startups continue to face uncertainty, with prolonged timelines for regulatory approval in the EU delaying market entry and potentially stalling business growth. It is only now, that we are getting to the point where there are might be some light at the end of the tunnel. However, perhaps more concerningly in the short-term term, the whole system (rightly or wrongly) has developed a reputation for delays and a long lead-time on European regulatory approvals.
FDA Predictability
In contrast to the evolving landscape in the EU, the FDA offers well-established pathways for medical device approval, including the 510(k) Premarket Notification and the Premarket Approval (PMA) process. Moreover, the FDA has implemented expedited review programs such as the Breakthrough Devices Program and the Priority Review designation, aimed at accelerating the approval process for innovative technologies addressing unmet medical needs. The regular publication of new guidance, particularly in evolving technology such as cybersecurity and AI has (in contrast to the EU) helped to establish the FDA as a much more proactive regulator during the same period that the EU has stumbled.
This has led to an emerging perception amongst medical device companies and investors that the FDA offer far greater predictability and faster turn-around times for review when compared to the regulatory environment in the EU. All of this, while still maintaining its global reputation as a robust and rigorous regulator.
Larger Market, Investor Preference and Market Perception
The US remains the single largest and most lucrative market for medical devices globally. Although a US strategy was always important to investors it is now vital and needs to be foremost in the business plan of most medical device start-ups.
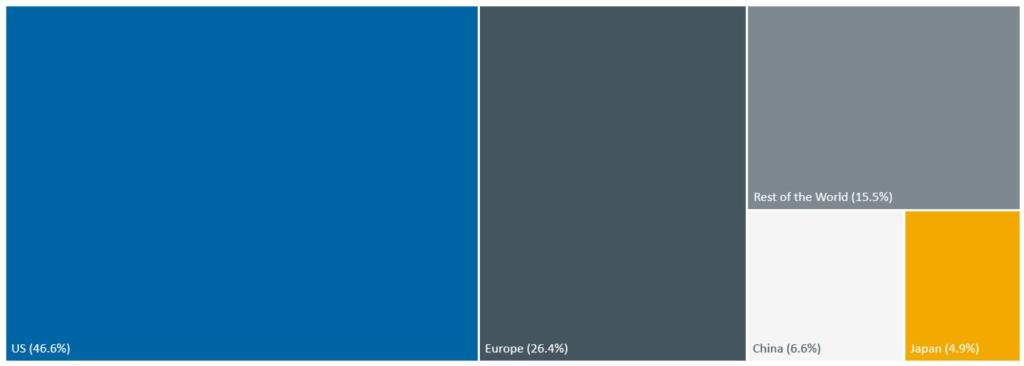
Source: MedTech Europe calculations based on the World Medical Device Factbook 2023 by Fitch Solutions. (Manufacturer prices, medical technology excluding IVD) From <https://www.medtecheurope.org/datahub/market/#sources>
Launching in the US first allows companies to establish a foothold in a market with greater commercial potential, enabling them to generate revenue and continue to attract investment to support further expansion. Additionally, securing FDA approval may facilitate reimbursement negotiations with private payers and health insurers, streamlining market access and providing an established path to sustainable revenue streams.
The preference among investors for startups prioritising a US strategy is therefore well-established. For medical device startups seeking funding to support product development and commercialisation efforts, prioritising FDA approval can enhance credibility and attractiveness to investors.
Concerningly, the regulatory challenges outlined above appear to have diminished the weighting given to a start-up's EU market strategy. It's not that the EU is no longer important or that it doesn't still represent a sizeable, valuable market, but rather the delays and uncertainty around timescales make it less attractive for investment. Startups still need to have an EU strategy, but it's not the thing that's going to secure investment. First and foremost, investors want to know what the US strategy is going to be.
Furthermore, securing FDA approval and launching in the US market first can bolster the perception of a company's brand and product quality, paving the way for successful market penetration both domestically and internationally.
Conclusion:
The decision of where to seek regulatory approval for medical devices carries significant implications for startups. While the EU offers a sizeable market and opportunities for innovation, delays and uncertainties in the regulatory approval process have prompted many startups to prioritise FDA approval and launch in the US market first. The established pathways, expedited review programs, and market access opportunities offered by the FDA provide startups with greater predictability and commercial potential, facilitating early market entry and business growth.
As an ISO 13485 accredited medical device development consultancy, eg technology have a deep understanding of the unique challenges and regulatory requirements of the MedTech industry, including FDA compliance, CE marking, and UKCA marking. At eg technology, we have supported many of our clients in successfully navigating their way to FDA approval. Our development process ensures you have the documentation needed for your design history file and we keep ourselves up to date with the latest guidance to give your product the best possible chance at securing that all important regulatory approval.
For more information on getting your technology through regulatory approval across markets, or to chat with one of the eg team about your product design and development requirements, please get in touch:
Via email: design@egtechnology.co.uk, by giving us a call on +44 01223 813184, or by clicking here.