Cresset, innovative provider of solutions for CADD, DMTA efficiency and contract research, releases Flare™ V7, a comprehensive drug design platform that integrates the power of ligand and structure-based methods.
New features and methods in Flare V7 include significantly enhanced Dynamics and Flare FEP calculations, a new ensemble docking method, enhanced quantitative SAR (QSAR) and Quantum Mechanics (QM) methods, and more tools to troubleshoot Flare FEP experiments, analyze dynamics trajectories, and prepare ligand structures for further studies.
“The new and enhanced methods in Flare V7 are based on our latest science and in response to feedback from our customers of the software, ensuring that we deliver what the users require to accelerate their drug discovery projects,” said Giovanna Tedesco, Head of Products, Cresset. “Developments in our underlying scientific methods deliver efficiencies and enable researchers to work collaboratively in an easy-to-use environment.”
The Flare V7 release includes:
-
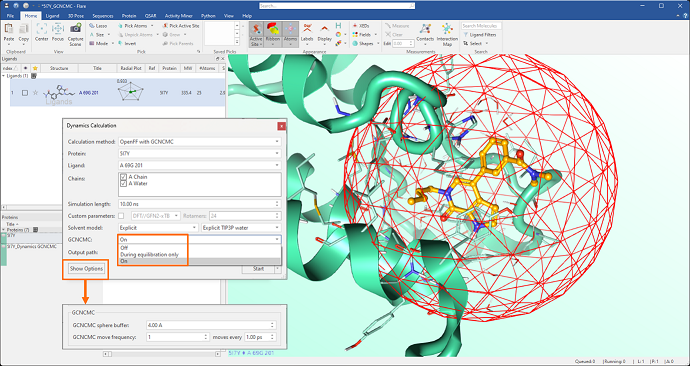
Introduction of GCNCMC (Grand Canonical Nonequilibrium Candidate Monte Carlo) in Dynamics and Flare FEP calculations to improve the sampling of hydration patterns into the protein active site.
-
Merging Covalent Docking and Ensemble Docking to create the new Ensemble Covalent Docking workflow, enabling users to consider active site flexibility when generating binding poses for covalent inhibitors, by including multiple protein active site conformations within the same docking experiment.
-
Expanded choice of methods for performing QM calculations on larger ligands, through the inclusion of the computationally efficient GFN2-xTB semi-empirical method
-
Wider range of methods for building predictive QSAR models, now including Consensus regression and classification, enabling Flare to predict the activity of new compounds based on the consensus of predictions from key regression and classification models


